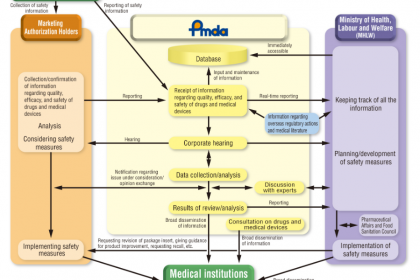
在全球醫療器械監管體系中,日本憑借嚴謹的監管架構、規范的注冊流程、完善的質量管控及專業化的執行機構,構建了覆蓋醫療器械“上市前注冊-上市后監督”全生命周期的監管體系。日本醫療器械監管以厚生勞動省(Ministry of Health, Labor and Welfare,MHLW)為統籌核心,其管轄的獨立行政法人藥品和醫療器械綜合機構(Pharmaceuticals and Medical Devices Agency,PMDA)承擔具體技術審評、審核及市場監督等職能。本文系統梳理日本醫療器械核心監管機構(PMDA與MHLW)的職能分工、全流程注冊要求(重點適配外國制造商)、質量管理體系規范及上市后監督要求,深入解析各環節核心要點,為醫療器械企業(尤其是外國企業)拓展日本市場、合規開展注冊申報及運營提供、、系統的行業參考(本文所指醫療器械不含體外診斷試劑)。
一、核心監管機構解析:PMDA與MHLW的職能分工與協同機制
日本醫療器械監管體系以“MHLW統籌管控、PMDA專業執行”為核心架構,兩者權責清晰、協同聯動,同時明確各類參與主體的監管要求,共同保障醫療器械的、有效與質量可控,為醫療器械注冊與運營提供堅實的監管支撐。
(一)厚生勞動省(MHLW):統籌全局的主管部門
MHLW作為日本醫療器械監管的主管部門,核心使命是保護日本民眾免受因藥品和醫療器械不、無效或質量不達標造成的健康危害,聚焦宏觀統籌、法規制定與最終審批,具體職能包括:
- 制造商注冊管理:負責統籌醫療器械制造商(含日本本土及外國制造商)的注冊審批與管理,規范制造場所合規性;
- 市場參與者許可:對醫療器械上市許可持有人(MAH)、經銷商、維修服務提供商等核心參與者進行許可管理,明確準入標準;
- 法規與標準制定:頒布醫療器械相關部級法令、指導方針及行業標準,明確監管實施細則與技術要求(如第169號部令MO 169);
- 最終授權發布:作為醫療器械上市授權的最終決策部門,審定PMDA提交的審評結果,發布醫療器械上市授權;
- 機構監督管理:對PMDA的工作進行監督與指導,確保其技術審評、審核及市場監督工作合規、開展。
(二)藥品和醫療器械綜合機構(PMDA):專業執行的核心技術機構
PMDA全稱為Pharmaceuticals and Medical Devices Agency,是MHLW管轄下的獨立行政法人,成立于2004年,專注于醫療器械、藥品、再生醫學產品等的具體監管執行工作,是日本醫療器械注冊與監管的核心技術支撐機構,核心職能包括:
- 授權程序執行:負責醫療器械上市授權的具體技術審評工作,開展監管審查,為MHLW的最終授權提供專業技術支撐;
- 專業咨詢服務:就醫療器械目標臨床研究方案、上市授權申報等事宜,為企業提供專業咨詢與指導,助力企業合規申報;
- GMP合規檢查:對醫療器械制造商(含本土及外國)開展良好生產規范(GMP)檢查,核查制造場所與生產過程的合規性;
- 市場監督管控:承擔醫療器械上市后市場監督工作,跟蹤產品使用,處置不良事件與質量隱患;
- 質量管理體系審核:負責高風險醫療器械及部分中等風險醫療器械的質量管理體系(QMS)審核,確保企業質量管控達標。
(三)PMDA與MHLW的協同機制
PMDA隸屬于MHLW,接受MHLW的監督與指導,兩者形成“宏觀統籌+專業執行”的協同模式:PMDA負責所有醫療器械的技術審評、GMP檢查、QMS審核、市場監督等具體專業工作,提出審評意見與審核結論;MHLW基于PMDA的專業意見,負責法規制定、參與者許可、制造商注冊統籌、最終授權發布等宏觀管控工作,確保監管工作的專業性、性與統一性。
二、日本醫療器械全流程注冊指南(重點適配外國制造商)
日本醫療器械注冊流程規范、環節清晰,核心遵循“質量管理體系搭建—指定授權代表—制造商注冊—產品分類—授權程序選擇—技術文檔提交—QMS審核—上市授權—上市后維護”的邏輯,其中外國制造商與日本本土制造商的注冊要求存在差異,重點環節如下(共8個核心步驟):
Step 01:建立符合要求的質量管理體系(QMS)
作為外國制造商,無需提供“本國批準”證明即可申請日本醫療器械上市授權,但必須滿足日本質量管理體系(J-QMS)要求,核心遵循2014年修訂的第169號部令(MO 169)。
關鍵要點:
- MO 169與ISO 13485:2003基本一致,企業持有的ISO 13485證書可作為滿足J-QMS要求的重要參考,降低審核難度;
- MO 169第3章包含額外要求,需企業(制造商)與上市許可持有人(MAH)共同遵守,包括文件保留期限、警戒程序要求,以及制造商與MAH之間的溝通規則;
- 質量管理體系需覆蓋產品設計、生產、檢驗、倉儲、銷售等全流程,確保產品質量可控。
Step 02:指定日本當地授權代表(MAH)
外國制造商在日本申請醫療器械上市授權前,必須指定一名日本當地的上市許可持有人(Marketing Authorized Holder,MAH),MAH是外國制造商進入日本市場的核心銜接主體,對醫療器械在日本的合規運營承擔全部責任,同時也是上市授權的所有者。
關鍵要點:
- MAH核心職責:不僅需位于日本、提交上市授權文件,還需對QMS承擔全部責任,負責醫療器械及批次放行、上市后市場監督、不良事件報告等工作;
- MAH資質要求:并非所有實體均可擔任MAH,需先向MHLW申請營業執照(稱為KYOKA),執照分為三類,對應不同的醫療器械類別授權范圍:
1. 類:可擔任所有類別(Ⅰ、Ⅱ、Ⅲ、Ⅳ類)醫療器械的MAH;
2. 第二類:可擔任Ⅰ類、Ⅱ類醫療器械的MAH;
3. 第三類:僅可擔任Ⅰ類醫療器械的MAH。
Step 03:完成外國制造商注冊(FMR)
在醫療器械獲得日本上市授權前,外國制造商需在MAH的協助下,向MHLW完成外國制造商注冊(Foreign Manufacturer Registration,FMR),所有負責產品開發、最終組裝或生產的制造場所均需注冊,組件制造商無需注冊。
關鍵要點:
- 注冊提交:通過Form 63-5提交注冊申請,需提供完整、準確的相關信息,包括制造商名稱及地址、所有擬上市設備清單、經理聲明、合規負責人信息、生產現場詳細信息等;
- 注冊周期:屬于行政審核程序,流程相對簡便,從提交申請到完成注冊約需30天;
- 注冊有效期與維護:注冊有效期為5年,到期后需及時更新;若制造商地址、聯系人等信息發生變更,需在30天內通知MHLW。
Step 04:對醫療器械進行分類
產品分類是日本醫療器械注冊的核心前提,需結合日本醫療器械術語集(Japanese Medical Device Nomenclature, JMDN)編碼及產品風險等級,明確產品所屬類別(Ⅰ類、Ⅱ類、Ⅲ類、Ⅳ類),不同類別對應不同的授權程序、審核機構與監管要求,具體分類標準如下:
- Ⅰ類(一般醫療器械):風險,如手術刀,監管方式為地方政府備案,無實質性審查;
- Ⅱ類(管制醫療器械):風險中等,如電子內窺鏡,需通過第三方認證機構(RCB)審核;
- Ⅲ類、Ⅳ類(高度管制醫療器械):風險較高或,如透析器、起搏器,需通過PMDA技術審評,由MHLW發布最終授權。
Step 05:選擇適配的上市授權程序
根據產品分類及特性,選擇對應的上市授權程序,核心分為兩類:
- 低風險產品(Ⅰ類):采用備案程序,完成地方政府備案后即可上市;
- 中高風險產品(Ⅱ、Ⅲ、Ⅳ類):采用審核/審評程序,Ⅱ類由RCB審核,Ⅲ、Ⅳ類由PMDA審評,審核/審評通過后由MHLW發布授權。
Step 06:編制并提交日語技術文檔
技術文檔是醫療器械上市授權審核/審評的核心依據,所有提交的文檔必須為日語,提交格式遵循IMDRF國際公認的STED格式,核心包含STED概要文件及以下8個附件:
1. 附件A:開發記錄(含先前設備版本、全球授權情況);
2. 附件B:產品規格(詳細技術參數、性能指標等);
3. 附件C:穩定性和保質期數據(驗證產品有效期);
4. 附件D:符合適用標準和基本原則的證明;
5. 附件E:性能測試數據(驗證產品性能達標);
6. 附件F:風險分析報告(識別產品潛在風險及防控措施);
7. 附件G:制造相關資料(生產過程、質量監督、工藝等);
8. 附件H:臨床數據(根據產品風險等級,提供相應的臨床試驗或臨床驗證數據)。
補充說明:文檔審核過程中,企業可與監管機構(PMDA/RCB)進行公開溝通,若監管機構有疑問,文檔審核預定持續時間不中斷,確保審核推進。
Step 07:申請質量管理體系(QMS)審核
醫療器械上市授權審核/審評通過前,需完成QMS審核,審核機構根據授權程序確定(PMDA或RCB),審核由MAH提交申請,覆蓋MAH的QMS及外國制造商注冊生產場所的QMS,對每個醫療器械系列執行獨立審核。
關鍵要點:
- 審核形式:分為現場檢查與文件審核,具體采用哪種形式,由監管機構根據企業ISO 13485認證情況、先前審核結果、不良事件報告、制造過程復雜性等因素確定;
- 文件審核核心內容:包括制造現場概覽、組織圖、質量管理手冊、質量管理文件清單、制造過程及驗證資料、MAH合同、警戒程序等;
- 特殊說明:日本自2015年起加入醫療器械單一審計計劃(MDSAP),接受MDSAP審計報告,通過MDSAP審計的制造商可避免重復審核,簡化審核流程。
Step 08:維護上市監管,確保持續合規
醫療器械獲得上市授權后,需進入常態化上市后維護階段,由MAH牽頭負責,嚴格遵循上市后監督要求,確保產品持續合規、有效,具體要求詳見本章第三節(上市后監督)。
三、日本醫療器械上市后監督要求
日本醫療器械上市后監督以MAH為核心責任主體,核心目的是跟蹤產品使用,及時處置不良事件與質量隱患,保障日本民眾健康權益,相關要求明確規定于“良好警戒慣例”條例(部長條例135)中,核心要點如下:
- 不良事件報告責任:MAH有義務及時收集、整理醫療器械在日本市場使用過程中發生的不良事件,向制造商及監管機構(PMDA/MHLW)提交報告,不得遲報、漏報、瞞報;
- 警戒程序執行:MAH需建立完善的警戒程序,明確不良事件收集、評估、報告的流程與責任分工,確保快速響應、有效處置;
- 產品質量跟蹤:MAH需對上市醫療器械的質量進行持續跟蹤,定期開展質量回顧,及時發現并解決產品質量問題,必要時啟動召回程序;
- 資料留存與更新:MAH需按要求留存醫療器械上市后相關資料(如不良事件報告、質量回顧報告等),確保資料可追溯;同時及時更新制造商注冊信息、MAH信息等,確保合規性;
- 監管配合義務:MAH需積極配合PMDA/MHLW開展的上市后監督檢查,提供相關資料與數據,不得拒絕、阻礙監督檢查工作。
四、核心總結
日本醫療器械監管體系以“、有效、質量可控”為核心,形成了“MHLW統籌、PMDA執行、MAH牽頭、全流程管控”的完善生態。MHLW負責宏觀法規與最終授權,PMDA承擔專業技術審評與審核,MAH作為外國制造商的核心銜接主體,承擔著合規運營的主要責任。
對于外國醫療器械企業而言,想要順利進入日本市場,需重點把握三大核心:一是搭建符合MO 169要求的質量管理體系,充分利用ISO 13485與MDSAP的優勢簡化審核流程;二是選擇具備對應資質的MAH,規范完成外國制造商注冊與產品分類;三是嚴格遵循全流程注冊要求,規范提交技術文檔,同時重視上市后監督,確保持續合規。
本文系統梳理的監管機構職能、注冊流程、QMS要求及上市后監督規范,可為相關企業與從業者提供的參考,助力其完成合規申報與運營,把握日本醫療器械市場的準入要點與監管趨勢。