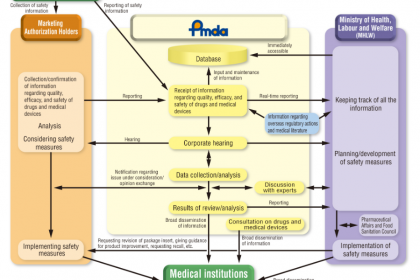
在全球醫療器械監管體系中,日本憑借嚴謹的分類標準、清晰的監管層級及專業化的審評機構,構建了覆蓋醫療器械全生命周期的監管框架。日本將醫療器械依據風險等級實施差異化監管,核心依托《藥事法》確立監管準則,由厚生勞動省(以下簡稱“厚生省”)統籌管理,醫藥醫療器械局(Pharmaceuticals and Medical Devices Agency,簡稱PMDA)承擔核心審評審批職能,確保醫療器械的、有效與合規性。本文系統梳理日本醫療器械的分類標準、差異化監管方式,深入解析PMDA的機構定位、核心業務及審評要求,為相關從業者提供、的行業參考(本文所指醫療器械不含體外診斷試劑)。
一、日本醫療器械分類標準:基于風險等級的分級體系
日本醫療器械的分類核心遵循“風險等級適配監管強度”原則,依據產品風險等級由低到高,明確分為三大類、四個等級,各類別界定清晰、適用范圍明確,為后續差異化監管提供基礎依據。
具體分類如下:
1. 一般醫療器械(Ⅰ類):風險等級,主要為結構簡單、性高,對人體健康無顯著潛在風險的醫療器械,無需復雜的性驗證即可保障使用,監管方式以備案為主。
2. 管理醫療器械(Ⅱ類):風險等級中等,產品結構與功能相對復雜,存在一定潛在風險,需通過針對性的認證或審評確保有效性,監管方式分為第三方認證與PMDA審評、厚生省承認兩種。
3. 高度管理醫療器械:風險等級,涵蓋Ⅲ類、Ⅳ類產品,此類產品技術難度高、直接影響人體生命,對有效性要求,核心監管方式為PMDA審評、厚生省承認,特殊情況下可采用第三方認證。
截至2018年4月1日,日本厚生省已制定946個認證基準、45個承認基準和9個審查指導原則,為各類醫療器械的分類界定、監管實施提供了明確的標準依據,確保分類與監管的科學性、統一性。
二、日本醫療器械差異化監管方式:基于分類的分級管控
依據2013年11月正式實施的《藥事法》,日本對不同風險等級的醫療器械采取“分級管控、施策”的監管方式,核心分為產品備案、第三方認證、厚生省承認三種,明確區分適用場景,兼顧監管效率與保障。
(一)一般醫療器械(Ⅰ類):產品備案制
一般醫療器械因其風險等級低,實行簡化監管,由備案人向PMDA提交備案申請,提交相關產品信息、性說明等資料,完成備案后即可上市銷售。備案制的實施,既降低了低風險醫療器械的上市門檻,又通過PMDA的備案審核,確保產品基本合規。
(二)管理醫療器械(Ⅱ類):第三方認證與PMDA審評結合
管理醫療器械的監管方式根據認證基準是否明確分為兩種情形:
1. 有明確認證基準的,由第三方認證機構開展認證工作(截至2016年11月30日,日本共有13家具備資質的第三方認證機構),認證通過后即可上市;
2. 無明確認證基準或產品不符合現有認證基準的,需由PMDA開展技術審評,審評通過后報厚生省進行承認,獲得承認后方可上市。
(三)高度管理醫療器械(Ⅲ類、Ⅳ類):以厚生省承認為核心
高度管理醫療器械直接關系人體生命,監管最為嚴格,核心監管流程為PMDA審評、厚生省承認。同時,針對已有明確承認基準和審查指導原則的高度管理醫療器械,也可由第三方認證機構開展認證工作;若申請認證的產品不符合承認基準或審查指導原則,則需重新向厚生省提交承認申請,經PMDA審評、厚生省承認后,方可上市。
三、PMDA核心解析:日本醫療器械監管的核心執行機構
從日本醫療器械的分類監管流程可見,PMDA作為厚生省醫藥食品局管轄的獨立行政法人,是日本醫療器械監管體系的核心執行機構,承擔著審評審批、管控、健康損害救濟等關鍵職能,成立于2004年,其工作成效直接決定日本醫療器械的監管水平與產品質量。
(一)PMDA機構概況
PMDA隸屬于厚生省醫藥食品局,為獨立行政法人,專注于醫療器械、藥品、再生醫療產品等的監管相關工作。截至2017年4月1日,PMDA共有員工812名,其中審查部門575名、對策部門198名、健康損害救濟部門39名;據2014年4月公布的數據,PMDA專門負責醫療器械審查的人員達104名,為醫療器械審評工作提供了充足的專業人力支撐。
(二)PMDA核心業務板塊
PMDA的業務核心圍繞“風險管控、保障”展開,涵蓋審查、對策、健康損害救濟三大板塊,其中審查業務是其針對醫療器械監管的核心職能,具體如下:
1. 審查業務(核心職能)
審查業務聚焦醫療器械上市前的有效性審核,同時覆蓋上市后再審查、生產環節監管等,核心職能包括:
- 臨床試驗相關咨詢,為醫療器械臨床試驗的開展提供專業指導;
- 醫療器械(含新器械、改良器械、仿制器械)的上市前審查、再審查及再評價;
- 申請資料可靠性調查,開展GCP(臨床試驗質量管理規范)、GLP(非臨床研究質量管理規范)、GPSP(良好藥品流通規范)符合性評估,確保申報資料真實、合規;
- 生產企業監管,開展GMP(藥品生產質量管理規范)、QMS(質量管理體系)、GCTP(良好臨床試驗用醫療器械質量管理規范)檢查,規范企業生產行為;
- 注冊認證機構檢查,對具備醫療器械認證資質的第三方機構進行監管,確保認證工作合規、嚴謹;
- 標準制定,參與日本藥典等相關標準的編寫與調查工作,完善醫療器械監管標準體系。
2. 對策業務
聚焦醫療器械上市后的監測與風險管控,跟蹤產品上市后的使用情況,及時發現并處置產品隱患,防范事故發生,保障使用者健康權益。
3. 健康損害救濟業務
針對因使用醫療器械、藥品等產生的健康損害,提供救濟服務,規范救濟流程,保障受害者的合法權益。
(三)PMDA醫療器械承認審查分類及要求
PMDA開展的醫療器械承認審查,根據產品類型分為新醫療器械、改良醫療器械、仿制醫療器械三類,各類別審查要求差異化明確,確保審查的性與針對性:
1. 新醫療器械:與已批準的醫療器械在結構組成、使用方法、效果及性能方面有明顯差異的產品,此類產品無明確審查標準,無論風險等級為Ⅱ、Ⅲ還是Ⅳ級,均由PMDA進行技術審評,審評通過后報厚生省承認。
2. 改良醫療器械:不屬于新醫療器械或仿制醫療器械的產品,此類產品通常無明確審查標準,需由PMDA開展技術審評,經厚生省承認后上市。
3. 仿制醫療器械:與已批準的醫療器械在結構組成、使用方法、功能、效果及性能等方面具有等同性的產品,即本質上與已批準產品一致,申請認證或承認時無需提供臨床試驗數據。自2009年起,已有明確審查標準的仿制醫療器械,可由第三方認證機構認證;無審查標準的仿制醫療器械,仍需由PMDA審評、厚生省承認。
(四)PMDA再審查要求
根據日本新版《藥事法》規定,初次獲得批準的醫療器械,需在上市后經過一定時間開展再審查,確保產品長期使用的有效性,具體再審查時間要求如下:
- 新設計、結構新穎或采用新原理的醫療器械,在獲得初次批準后第四年接受再審查;
- 具有新效力、新用途或新性能的醫療器械,在獲得初次批準后第三年接受再審查。
四、核心總結
日本醫療器械監管體系以“風險分級、管控”為核心,通過清晰的分類標準、差異化的監管方式,實現了對不同風險等級醫療器械的全流程監管,既保障了產品有效,又兼顧了行業發展效率。PMDA作為核心執行機構,憑借專業化的審評團隊、完善的業務體系,承擔著審評審批、管控等關鍵職能,與厚生省、第三方認證機構協同,構建了嚴謹、的監管生態。
了解日本醫療器械分類標準、監管方式及PMDA核心職能,對于醫療器械企業拓展日本市場、合規開展生產經營活動,以及相關從業者掌握行業監管動態,具有重要的指導意義。